Data
Allele specific binding (ASB) data for various transcription factors were retrieved from the ADASTRA database (Abramov et al. 2021) and a eY1H experiment (Bass et al. 2015). Training was conducted on chip-seq data from ADASTRA source experiments. SNPeBoT in its current iteration makes its prediction solely on the effect of Nucleotides 7 basepairs up and downstream of the mutation (or the midpoint of a 51bp sequence as the case would be for insertions, deletions or multiple mutations), therefore all ADASTRA derived non-concordant ASB events were discarded to controll for off-site effects on ASB. After this filtration step 34374 data points remained for 219 Different Transcription Factors (TFs).
Further filtering out of data was conducted for datapoints where the relevant TF could not be assigned to a PBM motif. Upon balancing of loss and gain classes 7014 ASB remained, 3506 for loss and gain respectively. To add no change data (control SNPs) we took a further 3506 SNPs from the ADASTRA source experiments that had no significant ASB measured and added these to the total training set which was composed of 10518 data points.
The eY1H data, in full, was reserved for testing. After data processing, as conducted on the ADASTRA set, we retained a total of 52 ASB events from an initial set of 294. Unlike chip-seq experiments, which may be influenced by the context in which SNPs occur, any effect measured in eY1H experiments will be down to changes in a TFs affinity to the nucleotide sequence at the binding site.
Data Processing
A single SNP prediction has to undergo multiple levels of processing to yield the input features upon which the prediction is made. First the amino acid sequence of the TF acting upon the SNP is retrieved and compared against a database of motifs, for which PBM experiments have been performed, using BLAST (Altschul et al. 1990). Any motif with 70% or more sequence identity with the query TF is selected as a match. This preprocessing step was handled by SBILib (Gohl et al. 2023). Reference and Alternate sequences are retrieved from the UCSC genome browser (Raney et al. 2024) or genopyc (Gualdi et al. 2024) for VCF or RSID input format respectively.
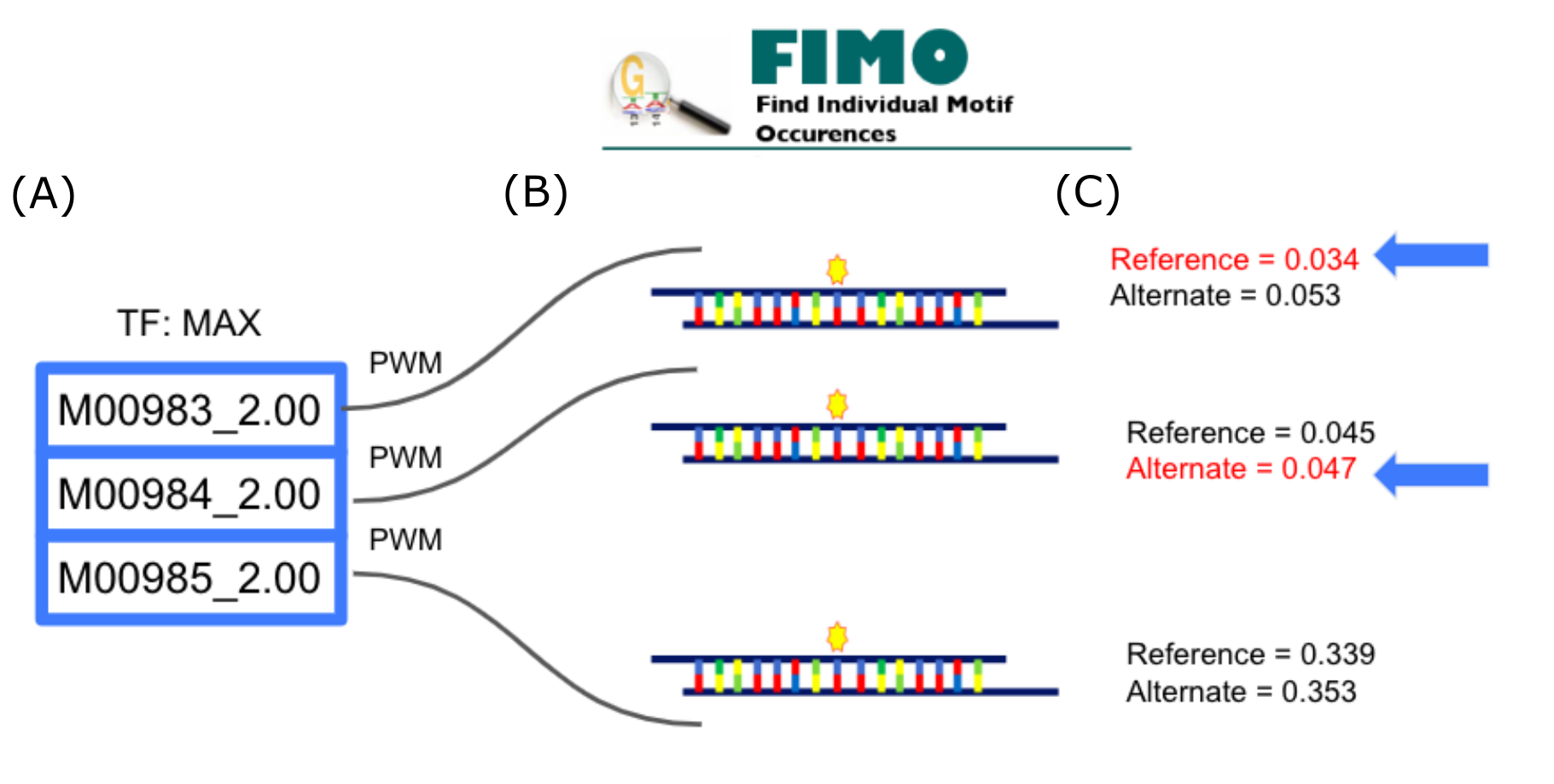
Subsequently the PWMs for these motifs are scanned against both the alternate and reference sequences using FIMO (Grant et al. 2011). For each sequence the motif with the best scoring FIMO hit incorporating the mutated position is selected as the binding motif Fig 1. For each sequence the relevant motif associated E-scores for all octomers incorporating the mutated position are retrieved, for a total of 16. Next a sliding window of average E-scores is assigned to each Nucleotide part of these octomers, 7 bases up and downstream of the mutated position, yielding a total of 30 E-score averages (15 for the reference and alternate sequences respectively). These 30 scores were then transformed into a 6x5 matrix.
Model
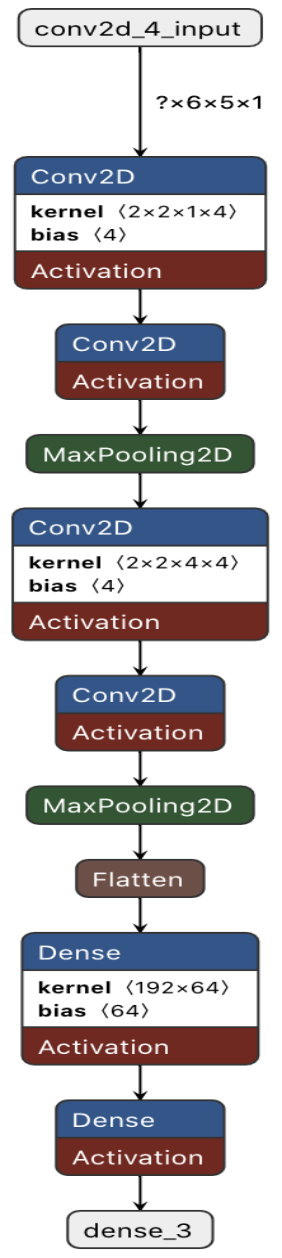
We trained a Convolutional Neural Network (Fig. 2) using Tensorflow (Abadi et al. 2016). The model takes an input (6,5,1) feature matrix. This is passed on two two initial 2D convolution layers and a pooling layer, followed by a second set of 2 convolution and 1 pooling layer. Finally the data is flattened and passed to 2 Dense layers with a final softmax activation function giving the model a 3 class output corresponding to the probability summing to 1 of each prediction “gain”, “loss”,”no change” in binding. To train the model our dataset was split into 70% training and 30% testing sets. From the training set a further split was made yielding a 10% validation set. All data was shuffled prior to the splits to ensure proper class representation.
Post-Hoc Control
In testing we saw a better performance of our model on Concordant Data. This was in accordance with our hypothesis that off-site effects would reduce SNPeBoT's ability to predict ASB events. This was further confirmed when applying the model to eY1H data where, at a threshold of 0.0001, an accuracy and coverage of 88 and 31% respectively was achieved. In order to account for cases where ASB was not solely due to alteration in DNA sequence at the binding site we implemented a Post-Hoc control method to ensure that there is an effect of the SNP on TF binding Fig 3.

Benchmarking
The same ASB data used for the post-hoc test was then applied to two established tools for prediction ASB, MotifbreakR (Coetzee et al. 2015) and atSNP (Zuo et al. 2015). Both tools are available as R packages and were installed with all necessary dependencies. For both MotifbreakR and atSNP we followed the provided vignette tutorials. In MotifbreakR each transcription factor had its motifs queried from MotifDB and predictions on the SNPs were made for these motifs scoring with the relative entropy method at a threshold of 1e-4. Because MotifbreakR doesn’t select a motif to apply to each SNP we frequently retrieved multiple predictions per SNP, corresponding to the various motifs for the given TF. To resolve this we looked at the allele difference for each prediction per SNP. When predictions were homogenous, favoring either reference or alternate allele entirely, the classification was either “loss” or “gain”. When predictions were discordant the SNP was classified as “no change”.
For atSNP each TFs publicly available PWMs from the CISBP database (Weirauch et al. 2014) were downloaded. Affinity scores and p-values and applied multiple testing adjustment using Storey’s q-value with the qvalue package (Storey et al. 2023) were calculated. We applied a qval_rank <= 0.05 filter to retrieve effect predictions. As with motifbreakR this left us with multiple predictions per SNP, reflecting the number of PWMs per TF, for which we employed the same method to retrieve a single prediction. Prediction accuracy and coverage were calculated for both MotifbreakR, atSNP and SNPeBoT. For the purpose of calculating performance metrics all cases resulting in a no change prediction were discarded.
Results
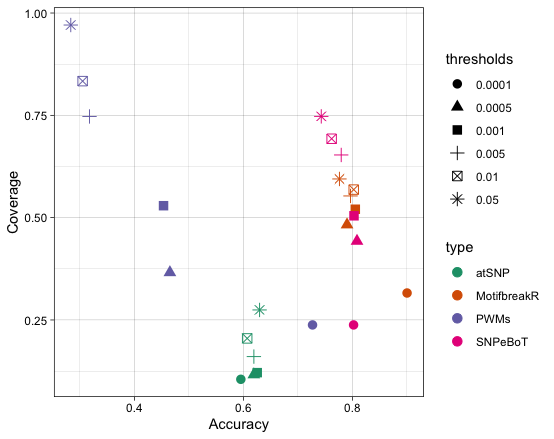
Holdout testing of the CNN model on Concordant Data achieved 80% accuracy at 100% coverage (2840 SNPs) with no distinction applied between the TF for which predictions were made. SNPeBoT prediction accuracy on eY1H data was 88% at 31% coverage. Results for Benchmarking Tests can be seen in Fig 4. SNPeBoT produces greater accuracy than a PWM only approach and atSNP. However, SNPeBoT accuracy remains lower than MotifbreakR in all but one threshold comparison. SNPeBoT is however capable of yielding higher percentage of coverage of ASB predicted on the TF tested.
Usage
Any submission must follow one of the three formats: VCF, Sequence, or rsid. We have provided examples and explanation to each fomat type on the Home page. For the webserver there are specific limits of SNPs that may be submitted for each Format type in the single TF prediction mode (Sequence:30, VCF:15, RSID: 1). In the Scanning prediction mode only a single SNP may be submitted for either format. For the eventuality that these limitations prove insuficient we have provided a standalone version of SNPeBoT at https://github.com/structuralbioinformatics/SNPeBoT. We suggest that for predictions on large numbers of SNPs or where many different TFs are to be run on the same set of SNPs that users install and run the standalone version.
References
1) Abadi, M., Barham, P., Chen, J., Chen, Z., Davis, A., Dean, J., ... & Zheng, X. (2016). {TensorFlow}: a system for {Large-Scale} machine learning. In 12th USENIX symposium on operating systems design and implementation (OSDI 16) (pp. 265-283).
2) Abramov, S., Boytsov, A., Bykova, D., Penzar, D. D., Yevshin, I., Kolmykov, S. K., ... & Kulakovskiy, I. V. (2021). Landscape of allele-specific transcription factor binding in the human genome. Nature communications, 12(1), 2751.
3)Altschul, S. F., Gish, W., Miller, W., Myers, E. W., & Lipman, D. J. (1990). Basic local alignment search tool. Journal of molecular biology, 215(3), 403-410.
4) Bass, J. I. F., Sahni, N., Shrestha, S., Garcia-Gonzalez, A., Mori, A., Bhat, N., ... & Walhout, A. J. (2015). Human gene-centered transcription factor networks for enhancers and disease variants. Cell, 161(3), 661-673.
5) Coetzee, S. G., Coetzee, G. A., & Hazelett, D. J. (2015). motifbreakR: an R/Bioconductor package for predicting variant effects at transcription factor binding sites. Bioinformatics, 31(23), 3847-3849.
6) Gohl, P., Bonet, J., Fornes, O., Planas-Iglesias, J., Fernandez-Fuentes, N., & Oliva, B. (2023). SBILib: a handle for protein modeling and engineering. Bioinformatics, 39(10), btad613.
7) Grant, C. E., Bailey, T. L., & Noble, W. S. (2011). FIMO: scanning for occurrences of a given motif. Bioinformatics, 27(7), 1017-1018.
8) Gualdi, F., Oliva, B., & Pinero, J. (2024). Genopyc: a python library for investigating the genomic basis of complex diseases. bioRxiv, 2024-01.
9) Raney, B. J., Barber, G. P., Benet-Pagès, A., Casper, J., Clawson, H., Cline, M. S., ... & Haeussler, M. (2024). The UCSC Genome Browser database: 2024 update. Nucleic Acids Research, 52(D1), D1082-D1088.
10) Storey JD, Bass AJ, Dabney A, Robinson D (2023). qvalue: Q-value estimation for false discovery rate control. doi:10.18129/B9.bioc.qvalue, R package version 2.34.0, https://bioconductor.org/packages/qvalue.
11) Weirauch, M. T., Yang, A., Albu, M., Cote, A. G., Montenegro-Montero, A., Drewe, P., ... & Hughes, T. R. (2014). Determination and inference of eukaryotic transcription factor sequence specificity. Cell, 158(6), 1431-1443.
12) Zuo, C., Shin, S., & Keleş, S. (2015). atSNP: transcription factor binding affinity testing for regulatory SNP detection. Bioinformatics, 31(20), 3353-3355.